|

Screening for would-be marriage partners
By Dhaneshi YATAWARA
Today the entire world focuses on an incurable deadly disease known
as Cooley's Anaemia. Never heard of it? Does the word Thalassaemia
strike a bell. Cooley's Anaemia is another name or Thalassaemia Major.
The word entered medical jargon after American paediatrician Dr. Thomas
B. Cooley, who described the disease in 1925.
Today is world Thalassaemia Day. 'Thalassa' in Greek is for the sea
and 'Haema' for blood. This easily preventable genetically transferred
disease is believed to have originated in the Mediterranean region and
today it is prevalent across the world largely affecting communities
bringing painful experiences specially to children. Today Thalassaemia
has become a major health problem for many developing countries as a
result of sticking to certain extreme traditional concepts.
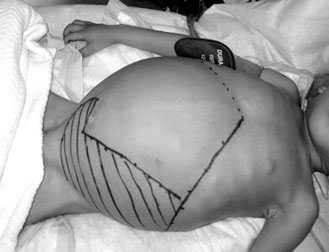 |
|
Thalassaemic child with
abdominal protrusion |
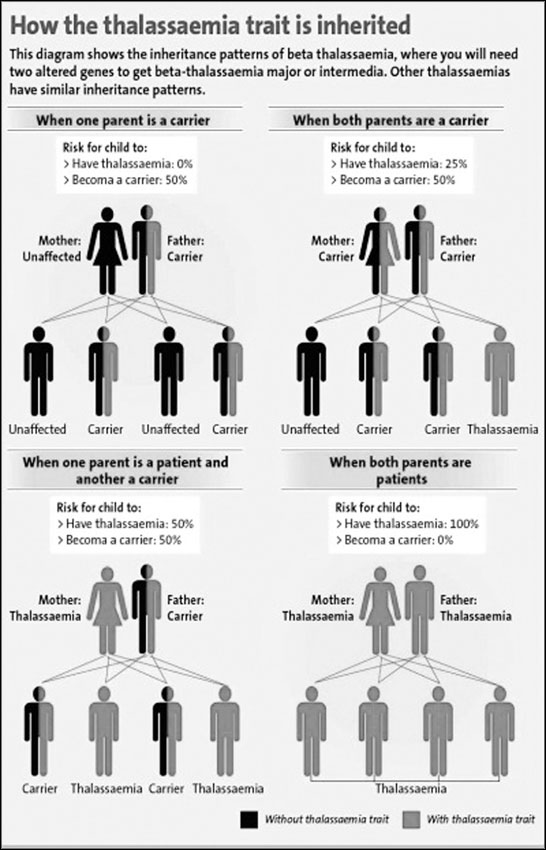
The only cause that spreads Thalassaemia is marriage between two
carriers. When marriage is between two blood relatives, the probability
is high for both the man and the woman to be carriers.
According to the Ministry of Health annually eighty children are born
with Thalassaemia Major. As of today the health sector has to look after
1,600 thalassaemia patients spending approximately Rs. 350,000 million
per year. The high risk areas in Sri Lanka are identified as
Northwestern, Uva, Northcentral and Western.
Thalassaemia is a disorder in the red blood cells. In these patients
red blood cells breakdown premature to its full life-span of 120 days.
The actual problem lies in the haemoglobin - the most important
constituent in blood carrying oxygen and giving blood its redness.
The Haemoglobin molecule has two components - haem and globin. Globin
is a protein. In Thalassaemia globin production gets defective as a
result of the changed genetic material. Haemoglobin is made of two
proteins: Alpha globin and beta globin. Alpha thalassaemia occurs when a
gene or genes related to the alpha globin protein are missing or
changed. Beta thalassaemia occurs when similar gene defects affect
production of the beta globin protein.
In both these types there are two forms - Thalassaemia Minor and
Thalassaemia Major. The individual suffering from Thalassaemia Minor has
only one copy of the defective gene together with one perfectly normal
gene. Persons with Thalassaemia Minor may almost have a slight lowering
of the haemoglobin level in the blood. Similar to a mild iron-deficiency
anaemia. However, persons with thalassaemia minor have a normal blood
iron level, unless they have iron deficient for other reasons. No
treatment is necessary for Thalassaemia Minor.
Patients with Thalassaemia Major has two defective genes for
thalassaemia and no normal gene. This causes a striking deficiency in
beta chain production and in the production of normal adult haemoglobin.
Thalassaemia Major is, therefore, a serious disorder.
In Sri Lanka
Since no one can predict the sequence of genes, Thalassaemia shows a
spectrum of conditions depending on the structure of the haemoglobin
molecule. "Only two have been observed in Sri Lanka - `beta'
Thalassaemia and `E' Thalassaemia," said Dr. Ashok Perera the Medical
Officer in Charge of the National Centre for Thalassaemia. Accordingly
patients with `beta' Thalassaemia is common. "Red blood cells in
Thalassaemia `E' patients shows longer life time - thus, less
complications for patients.
These patients live longer than patients with `beta' thalassaemia,"
he added. The oldest `beta' Thalassaemia patient living in the country
is 32-years-old and less number of Thalassaemia `E' patients live
longer.
According to Dr. Ashok Perera in `alpha' Thalassaemia the foetus gets
affected while in the mother's womb and either results in a miscarriage
or a still birth. "We don't have patients with `alpha' thalassaemia
since they do not live till birth," Dr. Perera explained.
Haemoglobin is not just one component - in our blood it has three
categories. i.e., haemoglobin A, haemoglobin A2 and haemoglobin F
(foetus). In a normal adult, haemoglobin A is in high percentage while
haemoglobin F is very low. Among normal Thalassaemia patients we mostly
find, haemoglobin F at a very high percentage and haemoglobin A is very
low. Haemoglobin F gets destroyed very quickly thus shortening the
lifespan of a red blood cell. "Hence, we cannot generalise patients.
They should be examined and analysed by a special consultant doctors and
a specially trained staff," Dr. Perera added.
Treatment
Treatment for Thalassaemia is still under research in the world.
Treatment for patients with Thalassaemia Major includes chronic blood
transfusion therapy, iron chelation, splenectomy, and stem cell
transplantation. In 2009, a group of doctors and specialists in Chennai
and Coimbatore registered the successful treatment of Thalassaemia in a
child using a sibling's (brother's) umbilical cord blood. The treatment
- a stem cell transplant has helped the girl to get rid of the disease.
According to news reports published on the web the stem cells were from
two sources - her brother's umbilical cord blood that was harvested
during the time of his birth and his bone marrow as the number of stem
cells from the cord blood was insufficient.
Since Thalassaemic patients experience anaemia, in order to fill up
the deficiency the body tends to produce more haemoglobin even at
extraordinary points. Normally red blood cells are produced in the bone
marrow at the end of long bones of our body. To meet the demand in a
Thalassaemia patient even maxillary bone marrow will start producing red
blood cells. The liver and the spleen are the other two organs that get
affected due to this excessive haemoglobin production. Hence the two
organs enlarge resulting a protruded abdomen. The facial and forehead
bones start to overgrow, trying to give more space for the bone marrow.
Hence, protrusions of the forehead and cheeks result.
A child would start showing these symptoms five years onwards.
 The initial symptoms becomes visible in a child since 4 - 5 months
from birth. "Lack of redness under the eye, inadequate weight gain, less
active, poor sucking, more prone to infections and eyes get yellow in
colour are - these are the most common ones. As the iron deposits under
the skins it becomes unusually dark in colour," Dr. Perera explained
further. The initial symptoms becomes visible in a child since 4 - 5 months
from birth. "Lack of redness under the eye, inadequate weight gain, less
active, poor sucking, more prone to infections and eyes get yellow in
colour are - these are the most common ones. As the iron deposits under
the skins it becomes unusually dark in colour," Dr. Perera explained
further.
To provide more space for bone marrow for maximum red blood cell
production the cavity enlarges and the shaft becomes thin. This results
in fragile bone structure.
Naturally these people have a high iron absorption rate to fill up
the deficiency as a natural adaptation of the body. "In addition to keep
haemoglobin at an optimum level monthly blood transfusion (2 - 3 days
per month) is required. This results in excessive level of iron deposits
in the body which needs to be removed using drugs," he added.
Iron is a foreign element to the body, thus it will have a negative
impact on the body's most vital organs. The heart tends to get enlarged
and later the patient maybe prone to heart failure. As the endocrine
system gets affected, i.e. organs like the pituitary gland, pancreas
etc. the body faces a hormone imbalance. With the irregular production
of insulin Thalassaemic patients are prone to diabetes. Secretion of
hormones affecting the secondary sexual growth imbalances, reducing the
growth of the child.
However, the final responsibility lies with the man and the woman who
are going to marry and produce children. Are we going to continue to
carry this burden to the next generation?
As this genetic disease is incurable and the cost of management is
very expensive, prevention is the best option for Sri Lanka. If a
married couple who are both carriers conceive a child, the couple has to
take the painful responsibility of raising a child with Thalassaemia,
restricting the right of the child to have a happy life. For a
successful prevention program, detection of all the carriers would be
ideal. In Sri Lanka if we promote to make sure that either of the
partners of a couple is a non carrier the concept will be more flexible.
As it is reiterated by medical professionals, the best option to
eradicate Thalassaemia is to prevent the birth of a Thalassaemia child.
To treat an adolescent patient the Government spends Rs. 1 - 1.5 million
per year per person. "People need to conduct blood screening, specially
if they are living in areas with high Thalassaemia prevalence. This is a
less complicated method," Dr. Perera said stating that it is a major
step in their prevention campaigns.
The first step would be testing for full blood count which could be
conducted at any medical lab islandwide, where the volume of red blood
cell is measured (MCV - Mean Corpuscular Volume). Three parameters are
considered - MCV, MCH (Mean Corpuscular Haemoglobin content ) and MCHC
and if these are below normal levels further testing is required to
differentiate patients from those having anaemic conditions. "A chemical
analysis of the haemoglobin is needed next specifically to identify
Thalassaemia patients and this is done through High Performance Liquid
Chromatography (HPLC)," Dr. Perera explained.
"A person must identify whether he or she is a carrier or not and
that should be a key factor when marriage is concerned. We call it the
21st `porondama' (horoscope factor)," Dr. Perera emphasised. This is a
test readily available at any main Government Hospital done free of
charge. For the Government it only costs Rs. 40 per test. After testing
the blood, a pink card is to be issued for thalassaemia carriers and a
green card to non-carriers. The Ministry of Health plans to issue
details to marriage registrars of high risk areas such as the North
Western, North Central and Uva Provinces. When two pink cardholders turn
up to get their marriage registered the registrar will educate them on
the risk of having children.
According to doctors two pink cardholders should avoid marriage or
having children. Blood Screening Centres are to be set up at the Jaffna
Teaching Hospital and Matara General Hospital to identify Thalassaemia
carriers in the respective areas. Such centres are already functioning
at all high risk areas.
The government's target is to reduce the number of children born with
thalassaemia to seven per year by 2015.
Bone deformity gene discovered
The Human Genetics team at The University of Queensland Diamantina
Institute have successfully used a new gene-mapping approach for
patients affected by severe skeletal abnormalities.
Skeletal dysplasias are a group of diseases that cause abnormalities
in the skeleton's growth and function.
This can lead to problems such as abnormal height and/or limb length,
difficulty with reproduction and decreased life span. Families affected
by skeletal dysplasias are usually very small in number, which can make
it difficult to find the disease-causing gene for that family.
Associate Professor Andreas Zankl, a clinical geneticist from The
University of Queensland Centre for Clinical Research, developed a Bone
Dysplasia registry for patients and their families - the first of its
kind in Australia.
Through the registry, the UQDI team of researchers met a family with
two young daughters affected by a severe form of dwarfism.
The team used next-generation sequencing to simultaneously study the
four immediate family members and compare their exomes - the coding
section of the genes - to each other and against the reference sequence
from the international Human Genome Project.
They were able to discover which gene within the family caused the
abnormality. Impressively, the mapping process took only a few weeks.
The UQDI researchers then successfully determined how the genetic
abnormality caused the skeletal disease.
In the past, researchers could only sequence and compare a few genes
at a time, which was expensive and time-consuming.
For example, UQDI researchers had spent a decade finding the
responsible gene for another type of skeletal dysplasia, fibrodysplasia
ossificans progressiva.
In contrast, next-generation sequencing technology can provide more
rapid results for mapping genes in these particular types of diseases.
However, despite this breakthrough in progress, Associate Professor
Emma Duncan said it was still an intensive process.
"Typically, we all have a number of small genetic differences - we
find approximately 20,000 on average just in our coding regions when
compared with the Human Genome sequence - so it's still a very involved
process to work out which one is the disease-causing mutation," she
said.
"For this family, it's been a huge relief to find out why their
little girls have this devastating skeletal disorder, and understanding
the genetics has helped them in their planning for any future
pregnancies," said Professor Matthew Brown.
With the success of their next-generation sequencing approach, the
team have also researched another skeletal dysplasia case which involved
five unrelated individuals, comparing their exomes with each other and
with the Human Genome Project.
By examining just this small number of affected people, the
responsible gene has been identified. UQDI researchers will continue to
map unknown genes for skeletal dysplasias and for other likely
single-gene inherited diseases.
The paper has been published in the Public Library of Science (PLoS).
(Source: The University of Queensland Diamantina Institute)
Measuring medications' effects on the heart
A common component in webcams may help drug makers and prescribers
address a common side-effect of drugs called cardiotoxicity, an
unhealthy change in the way the heart beats. Researchers at Brigham and
Women's Hospital (BWH) have used the basic webcam technology to create a
tool to look at the effects of medications in real time on heart cells,
called cardiomyocytes. These findings were published in the journal, Lab
on a Chip.
Researchers developed a cost-effective, portable cell-based biosensor
for real time cardiotoxicity detection using an image sensor from a
webcam. They took cardiomyocytes, derived from mouse stem cells, and
introduced the cells to different drugs. Using the biosensor, the
researchers were able to monitor the beating rate of the cardiomyocytes
in real time and detect any drug-induced changes in the beating rates.
The technology provides a simple approach to perform evaluative
studies of different drugs effects on cardiac cells. Cardiotoxicity is a
significant problem in drug development, with more than 30 percent of
drugs withdrawn from the market between 1996 to 2006 related to cardiac
dysfunction. "Assessing the toxic effects of new drugs during the early
phases of drug development can accelerate the drug discovery process,
resulting in significant cost and time savings, and leading to faster
treatment discovery," said Ali Khademhosseini, PhD, of the Center for
Biomedical Engineering at the Department of Medicine at BWH.
"This technology could also play a role in personalized medicine,"
said Sang Bok Kim, PhD, a Research Fellow in the Renal Division at BWH.
"By first extracting somatic cells from patients which can be
reprogrammed to stem cells called induced pluripotent stem (iPS) cells.
Then these iPS cells can be differentiated into cardiac cells to be
studied, the biosensor can monitor the cardiac cells as they're
introduced to a medication, providing a glimpse at how the drugs may
affect the individual's heart, and thus shaping the treatment plan for
that person."
Monitoring cardiac cells in the past required using expensive
equipment that had a limited measurement area. This low cost biosensor
is compatible with conventional equipment but will enable reliable, yet
faster and more cost-effective studies.
"Our next goal is to combine our detection sensor with our microwell
arrays and perform screening studies of thousands of drugs to cardiac
cells simultaneously in a fast and reliable manner," said Dr.
Khademhosseini. (Source: Holly Brown-Ayers Brigham and Women's
Hospital)
|















")